下载 1.sratoolkit 知道数据的SRR号,使用prefetch命令进行下载,即可以下载单个数据,也可以进行批量下载。下载单个数据可直接使用prefetch SRRx...
 发简信
发简信
IP属地:广西
下载 1.sratoolkit 知道数据的SRR号,使用prefetch命令进行下载,即可以下载单个数据,也可以进行批量下载。下载单个数据可直接使用prefetch SRRx...
我们拿到的观测数据常常是有缺失值的,有时候基于数据分布是可以预测缺失值。有时候却是不能,比如缺失值太多,数据量太小。因此,计算相关性时需要考虑到缺失值的使用。在R中的基础函数...

单细胞分析不可避免的会涉及到鉴定不同细胞类型的功能或比较相同类型在不同状态下的功能差异,因此,功能分析在单细胞分析中相当重要. 功能分析的方法很多,这儿我们主要讲述3种功能分...

生存模型的构建方法包括:1. Lasso回归;2. Cox多因素回归;3. 随机森林;4. 支持向量机 可以把log_rank_test或cox筛选出的基因单独做模型构建,也...
Error in if (slev[1] == 0 & slev[2] == 1) return(D) :
missing value where TRUE/FALSE needed
TCGA数据差异分析后生存分析(批量单因素cox回归/Lasso筛选,多因素cox建模,时间依赖ROC曲线及KM plot可视化)测序上游分析系列: mRNA-seq转录组二代测序从raw reads到表达矩阵:上中游分析pipelinemiRNA-seq小RNA高通量测序pipeline:从raw r...

测序上游分析系列: mRNA-seq转录组二代测序从raw reads到表达矩阵:上中游分析pipelinemiRNA-seq小RNA高通量测序pipeline:从raw r...
可以分享代码吗?
TCGA数据差异分析后生存分析(批量单因素cox回归/Lasso筛选,多因素cox建模,时间依赖ROC曲线及KM plot可视化)测序上游分析系列: mRNA-seq转录组二代测序从raw reads到表达矩阵:上中游分析pipelinemiRNA-seq小RNA高通量测序pipeline:从raw r...

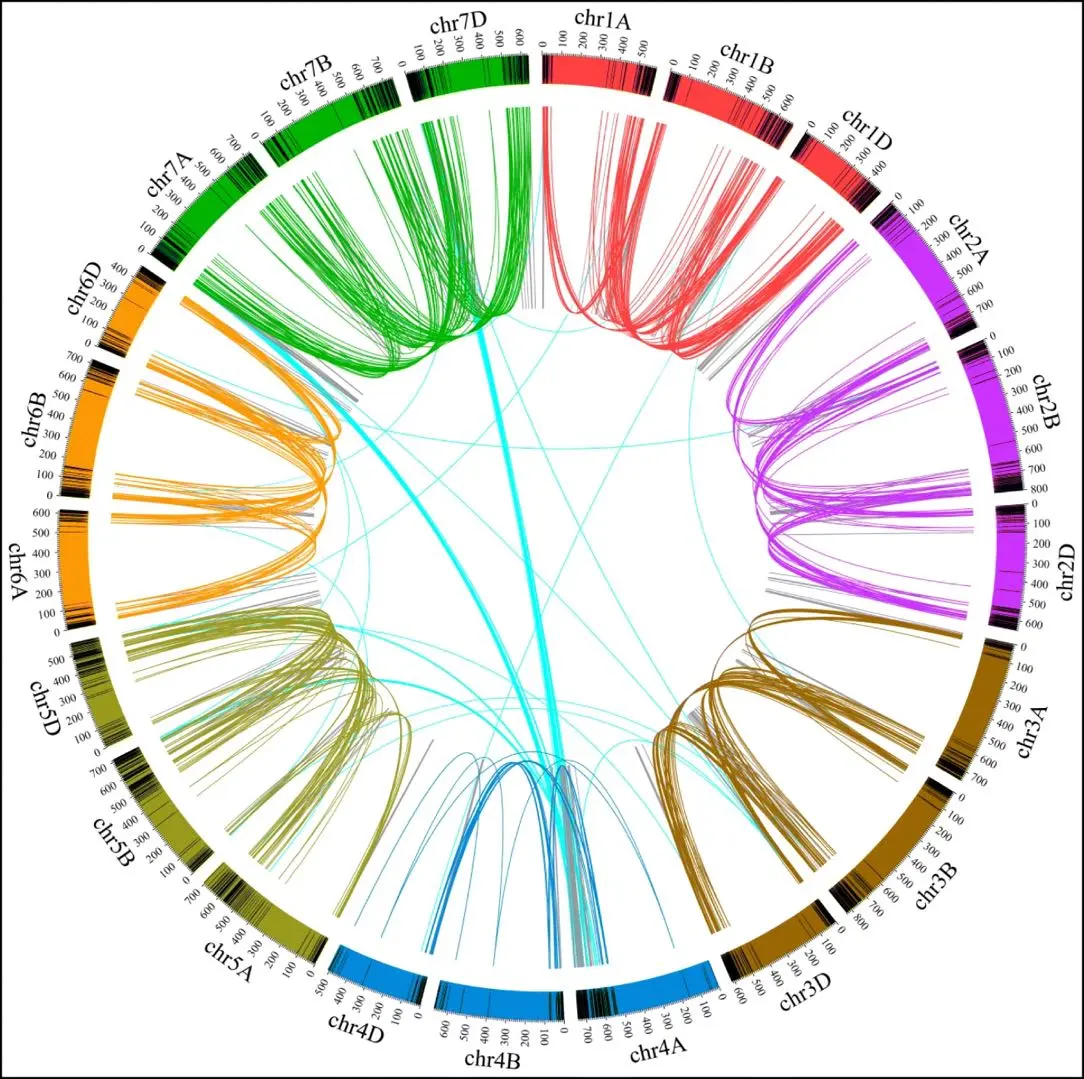
文献来源 doi: https://doi.org/10.1101/742320[https://doi.org/10.1101/742320]Title:A molecul...
您好,您有找到hg38的全长文件吗
生信小白教程之Count转TPM,FPKM相信很多科研工作者(不包括比我厉害的大佬们)在做转录组时,都是在公司做测序,然后数据也交给公司分析,又然后,期待做个差异分析,得出像下图那么完美的热图。 然而,然而,,我们得...
ScoreJackStra显示错误,下标出界
Seurat包基本分析实战一、背景知识 文献:https://www.aging-us.com/article/103695/text[https://www.aging-us.com/article...
遇到这种错误应该怎样解决:
Pipestance failed.Please see log at: SRR973158/SC_RNA_COUNT_CS/FULL_COUNT_INPUTS/WRITE_GENE_INDEX/foek0/chnk0-u726a8de7fe/_errors
Waiting 6 seconds for UI to do final refresh. Pipestance failed.
Use --noexit to keep UI running after failure
10X cellranger 报错解决办法越来越多的同学开始自己尝试做10X单细胞转录组数据分析了,除了Linux的基本命令之外,首先用到的软件是cellranger。失败是成功之母,对一个新手来讲很可能会遇到这样那...
您好,我想问一下,我从EBI下载的fastq文件,可不可以直接改名后进行cellranger count处理
cellranger count使用1、如果你是直接拿到的R1/R2的fastq文件,那么就直接上cellranger count。氮素,如果你是I1/R1/R2的数据,那就麻烦还要跑个cellranger m...