
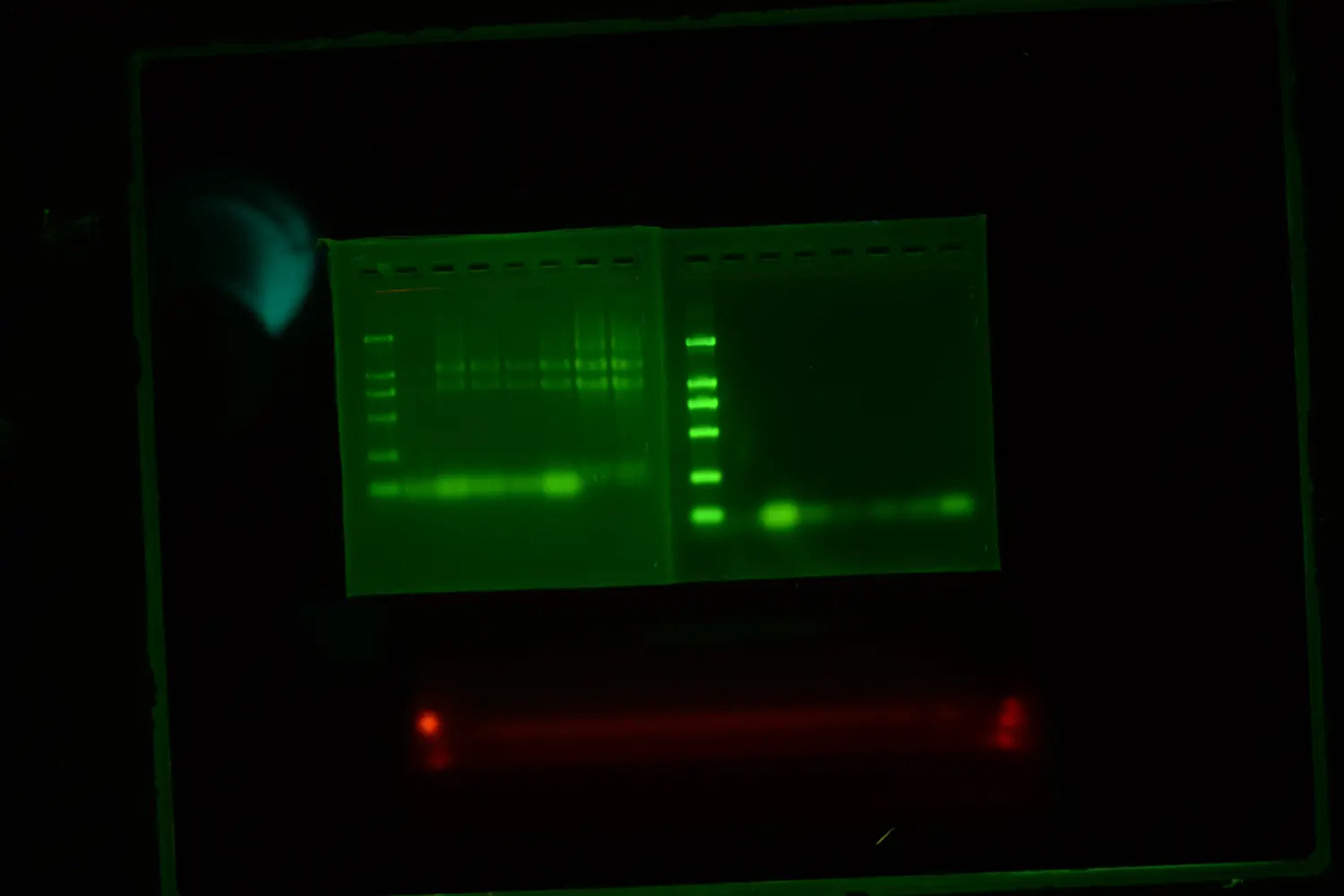
去年10月,我从做微生物方向的科研服务公司,跳槽到了一家准一流检验所新开的特检部门。听说老东家的微生物部门分崩离析,博士头头是去了肠道微生物相关的公司,骨干硕士去做癌症早筛了...
 发简信
发简信
IP属地:西藏
去年10月,我从做微生物方向的科研服务公司,跳槽到了一家准一流检验所新开的特检部门。听说老东家的微生物部门分崩离析,博士头头是去了肠道微生物相关的公司,骨干硕士去做癌症早筛了...
转录组Read Counts统计原理: 一般来说是通过比对工具(如HISAT2、STAR,fanse)将测序reads比对到参考基因组使用计数工具(如HTSeq、featur...
以下是对 samtools 各命令的详细中文解释及其拓展应用场景,结合功能分类和实际生物信息学分析需求整理: 一、索引命令(Indexing) dict功能:创建参考基因组的...
在开篇,我觉得应该首先和同学们说一下如题所示的问题。我发现到目前为止,可用于基因功能注释的网站还是有很多的,但绝大多数网站所收录的植物物种实在太少而植物各物种差别又太大。举个...

欢迎来到今日的《科学快报》 地球上和海洋中究竟有多少物种?——从DNA条形码看生物多样性 相关研究来源于PLOS Biology期刊。 你是否曾经好奇地想知道地球上和海洋中到...
今天,66大顺。祝大家科研顺利,万事顺心! psass用的人不多,所以觉得说不定可以分享一个文章,说不定有人感兴趣。 psass前几日安装一直不顺,没想到今日直接装成功了,都...

IQ-tree也是一款最大似然法构建进化树的软件,目前IQ-tree已经更新到2.0版本功能和性能也有很大的提升,主要有四大功能,高效建树(efficient tree re...

1安装方法 首先安装好java环境,通过官网下载最新版本的软件压缩包,然后解压即可,最好安装在自己熟悉的目录下。 java检测 java -version 配置数据库 有一些...

Hifiasm 使用教程 介绍 Hifiasm 是一个快速的单倍型解析 de novo 组装软件,最初设计用于 PacBio HiFi 读取。其最新版本可以通过利用超长的 O...

MG2C是一个很纯粹的工具。没有广告,没有推广,不考虑任何利益,纯属业余爱好。 MG2C是一个很“肤浅”的工具。打开使用界面,参数、输入和输出都在一个界面,看一眼界面就相当于...
不错,拿到MH的宣传册,里面看到你文章的宣传摘要,就来搜一下,看到作者了,谢谢你开发这个小公举😄
MG2C教程-点点鼠标就能完成画图MG2C是一个很纯粹的工具。没有广告,没有推广,不考虑任何利益,纯属业余爱好。 MG2C是一个很“肤浅”的工具。打开使用界面,参数、输入和输出都在一个界面,看一眼界面就相当于...

需要看下Nip和9311的基因匹配的有多少,思来想去,blast还是有点缺陷的,可能匹配到其他染色i他,需要用gff来帮助定一下位。所以,用基因的共线性分析看起来更合适一点。...
@顾大师 先配置好该配置的所有的,然后再装软件试试?
好久了,我也忘记了
2019-08-01 SMRTlink7.0 安装超不顺利。。。可参考安装流程:https://vcru.wisc.edu/simonlab/bioinformatics/programs/install/smrt.htm遇到问题1 查找...
1. 介绍 这个代码实现了一个名为Combat的类,该类用于数据的批次效应校正。Combat (ComBat) 是一种广泛应用于基因表达数据和其他高通量数据的批次效应校正方法...



