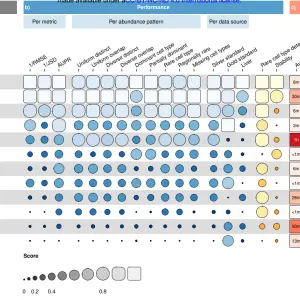
1、通过KEGGREST包来探索KEGG数据库的12大代谢通路 通过观察KEGG官网 :https://www.genome.jp/kegg/pathway.html[htt...
 发简信
发简信
IP属地:江苏

1、通过KEGGREST包来探索KEGG数据库的12大代谢通路 通过观察KEGG官网 :https://www.genome.jp/kegg/pathway.html[htt...
因为最终是cat打印到终端的,linux可以写个标准输出到文件中,R语言的话,建议改源代码,将其存为data.frame的形式
CORNAS:一种快速简单鉴定无重复转录组差异基因的方法还记得上次文章的最后提到CORNAS这种方法吗?最近刚好在Github上看到了这个项目,就花了点时间看了下文档感觉操作也比较简单,这里记录一下使用过程,大家共同学习一下。 介...

2021.02.14今天收集了三个与谱系分化有关的单细胞高级算法:MAGIC(2018 Cell),PHATE(2019 NBT),Palantir(2019 NBT)。它们...
理论 参考文章为:genesorteR[https://www.biorxiv.org/content/10.1101/676379v2.full]简单理解下,每个cell ...

关键词 FindTransferAnchors函数 TransferData函数 细胞类型注释 映射数据集/Transfer 数据集 适用背景 细胞注释的标准一直备受争议,就...
不同物种可参考《Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer》自行构建逻辑回归模型(弹性网络正则化)用于对齐不同的细胞类型
利用Seurat包Transfer数据集(FindTransferAnchors和TransferData)2022-6-09关键词 FindTransferAnchors函数 TransferData函数 细胞类型注释 映射数据集/Transfer 数据集 适用背景 细胞注释的标准一直备受争议,就...
https://github.com/satijalab/seurat/issues/3930 此问题的出现是由于细胞数过少产生的,IntegrateData运行过程中需要按照行或列取最小值生成一个对称的矩阵,默认是100。若细胞数小于100就会报错,将k.weight参数改为最小的细胞数就能成功运行了。
Error in idx[i, ] <- res[[i]][[1]] : number of items to replace is not a multiple of replacement ...调整下参数就好了
PLSDA是一种有监督的方法,可以用于计算两组差异的特征。用在这里比较不同样本的区别不太合适,还是得用pca
PLS-DA代替PCA试试降维效果?最近有小伙伴问道PLS-DA(Partial least squares Discriminant Analysis,偏最小二乘判别分析),一种组学分析中常用的多变量分析方法...
hello,大家好,周三了,一周又要过去了,时间真的是过得飞快,根本来不及反应,真的是一寸光阴一寸金,寸金难买寸光阴~~~ 今天分享一个小的知识点,单细胞关于巨噬细胞分析的常...

单细胞绘图系列: Seurat绘图函数总结[https://www.jianshu.com/p/95e61f7e834d] Seurat是分析单细胞数据一个非常好用的包,自带...
好的谢谢🙏
Scissor:联合表型数据,Bulk-seq和scRNA的分析软件前言 作者开发Scissor的目的是结合bulk-seq的数据,寻找与某一性状显著相关的单细胞亚群,然后从表型的角度解释这些细胞亚群的生物学意义。作者开发Scissor的动机...
作者您好有个问题,这里的bulk数据和单细胞数据必须对应同一来源的样本是吗
Scissor:联合表型数据,Bulk-seq和scRNA的分析软件前言 作者开发Scissor的目的是结合bulk-seq的数据,寻找与某一性状显著相关的单细胞亚群,然后从表型的角度解释这些细胞亚群的生物学意义。作者开发Scissor的动机...